 Hotline服务热线:010-61006450
Hotline服务热线:010-61006450
 简体中文
简体中文建议收藏!化学合成原料药杂质控制策略
ICH Q7A 对于原料药(Active Pharmaceutical Ingredient, API)的定义:
任何旨在供生产一种药物制剂,并作为其活性成分的物质或混合物,这类物质旨在疾病的诊断、治疗、缓减、处置和预防及影响人体的结构和功能等方面发挥药理作用或其他的直接效益。
根据原料药的合成工艺,结合药典(包括USP, EP, ChP, JP等)收载相关杂质对其中可能存在的有机杂质进行了理论分析和检测,确定是否订入标准进行常规控制。可能存在的有机杂质有:

重点:有机杂质研究范围需要包括所有药典(包括USP, EP, ChP, JP及其他法定标准,如进口注册标准,药品注册标准等)收载相关杂质,无论原料药工艺是否产生。
-
根据原料药的合成工艺,API分子结构,相关EMA审评报告,IF文件和化学机理分析初步确定降解杂质。
-
采用有关物质方法验证专属性-强制降解验,高温(高于加速10°C),高湿(RH92.5%),酸(0.1~1.0M)、碱(0.1~1.0M)、氧化(30%H2O2)、光照保证至少有一个条件下降解5%~10%左右,确定杂质降解途径。
稳定性期间增长,接近或大于鉴定限0.10%的降解杂质需要鉴定,并纳入API质量标准,进行常规控制;大于界定限0.15%降解杂质,尚未进行毒理学试验证明限度合理性,需要进行毒理学试验,证明安全性;或者优化结晶工艺,改善晶型、晶癖、粒径等因素,增强API的稳定性,不出现大于界定限0.15%的降解杂质。
开发有关物质相关方法检测上述有机杂质,杂质控制策略如下:

根据生产工艺和警示结构,确定产生致突变杂质,并依据ICH M7进行如下分类,确定相应的控制限度。

➯ 关注队列(比一般致突变性还毒)
已确定的某些结构基团具有较高的致癌性,即使摄入量低于TTC水平,理论上仍会具有高致癌风险,这类高效致癌性致突变致癌物,被称为关注队列,其中包括黄曲霉毒素类、N-亚硝基化合物、烷基-氧化偶氮基化合物。
➯ 致突变杂质
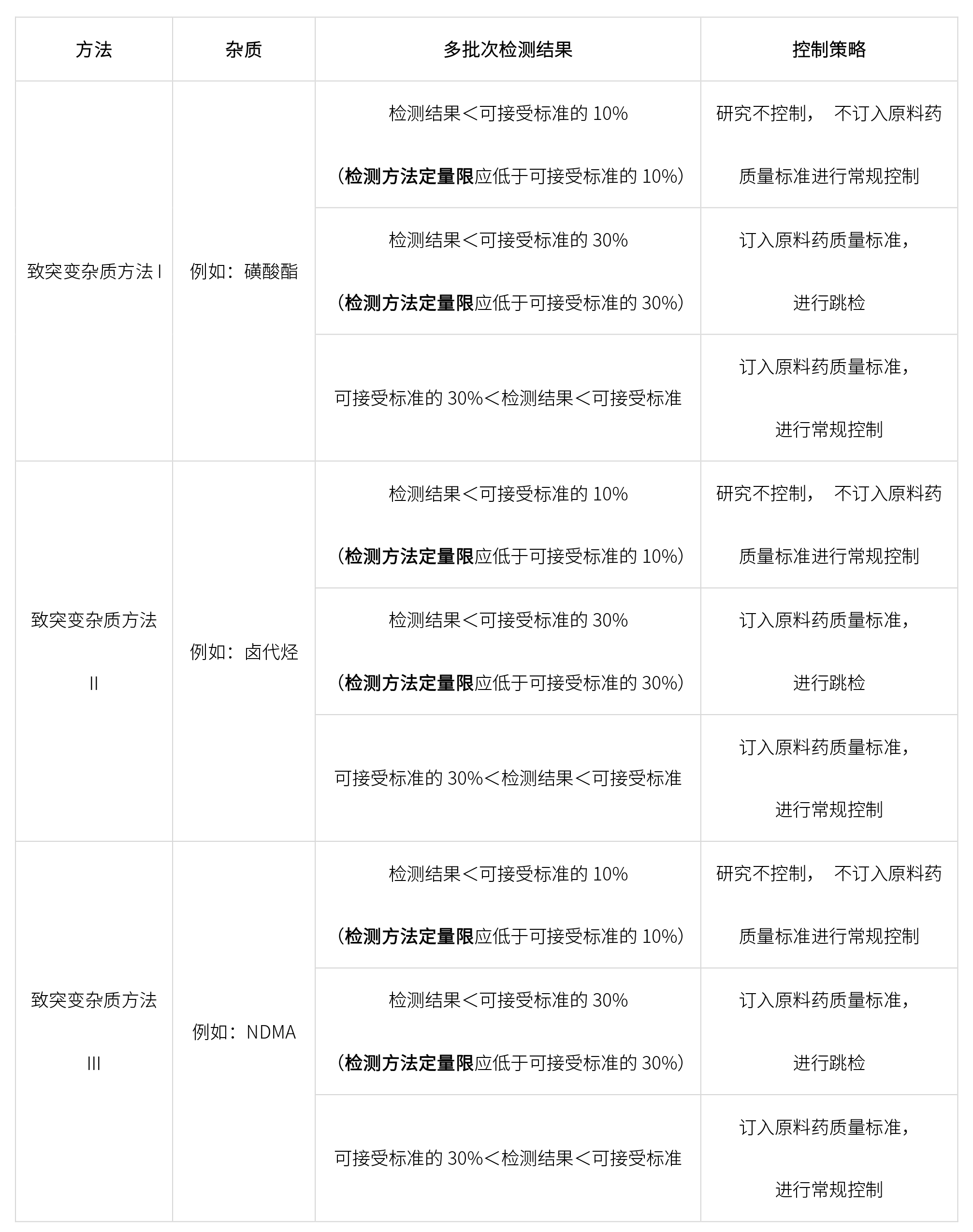
开发致突变杂质相关方法检测原料药中致突变杂质,杂质控制策略如下:
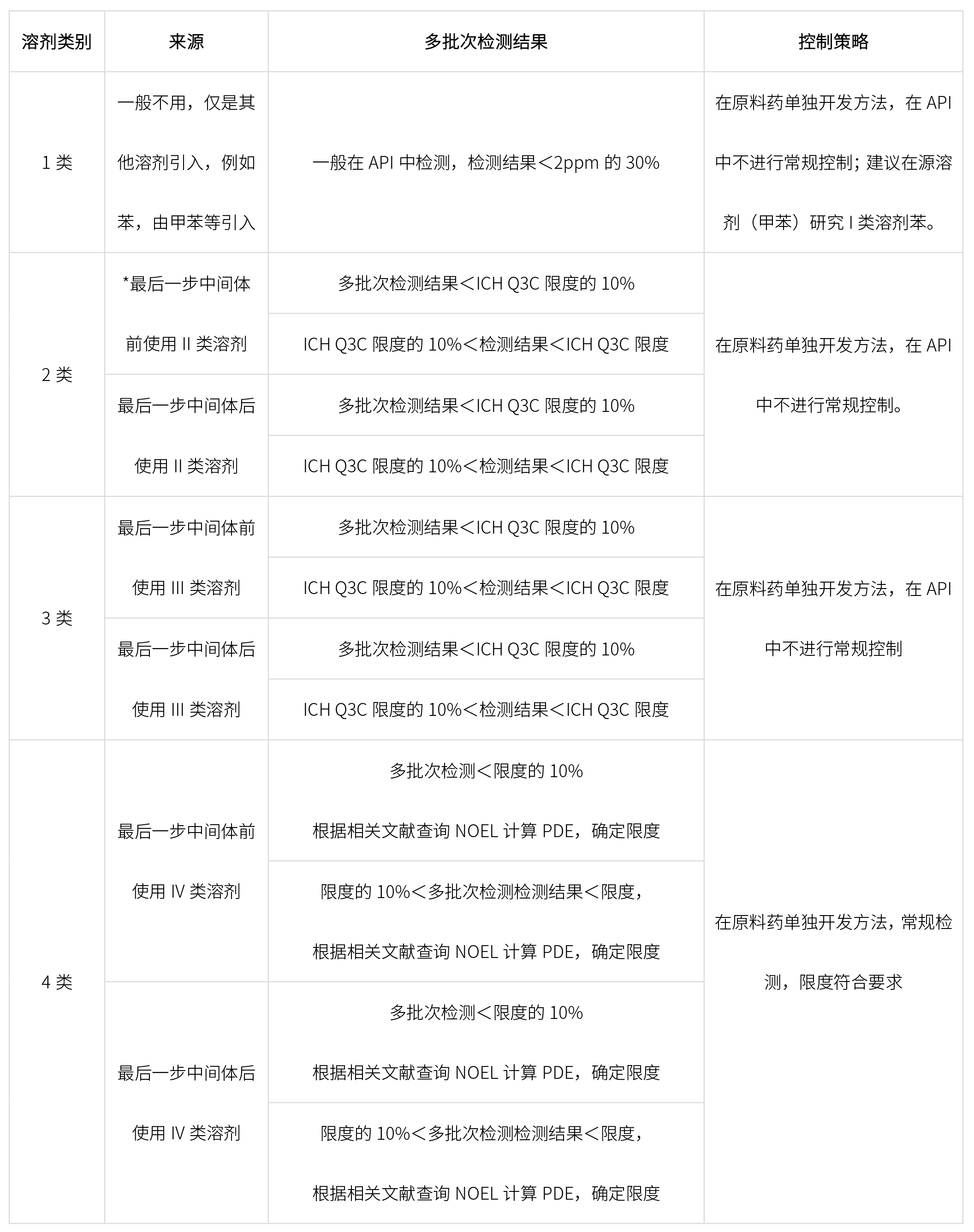
根据终产品合成工艺,结合ICH Q3C,确定控制策略如下:
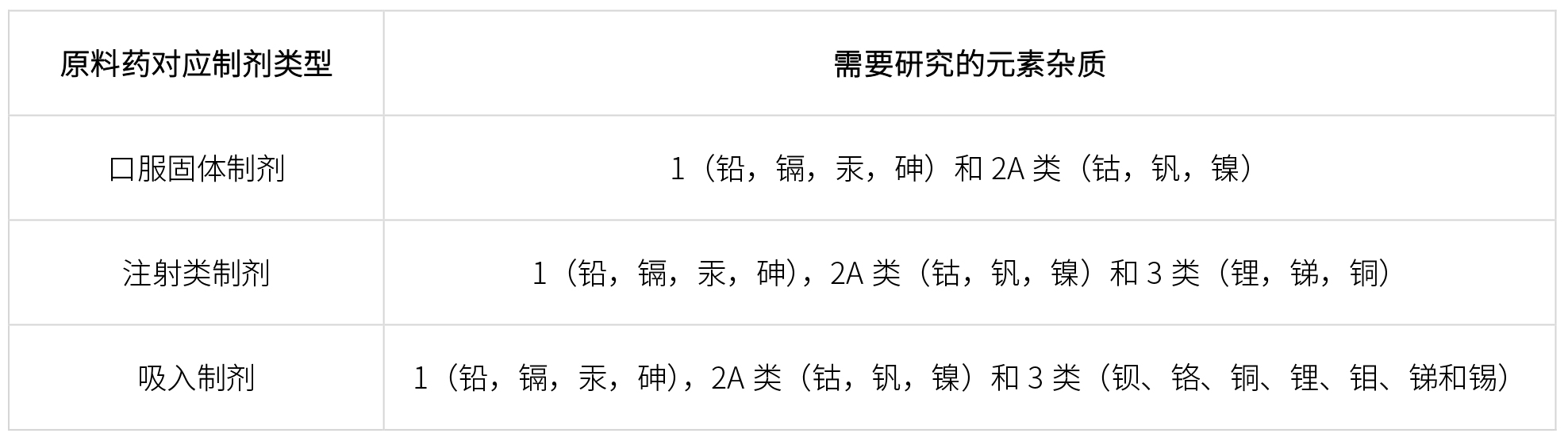
根据ICH Q3D指导原则,不同类型原料药至少需要研究如下元素杂质:
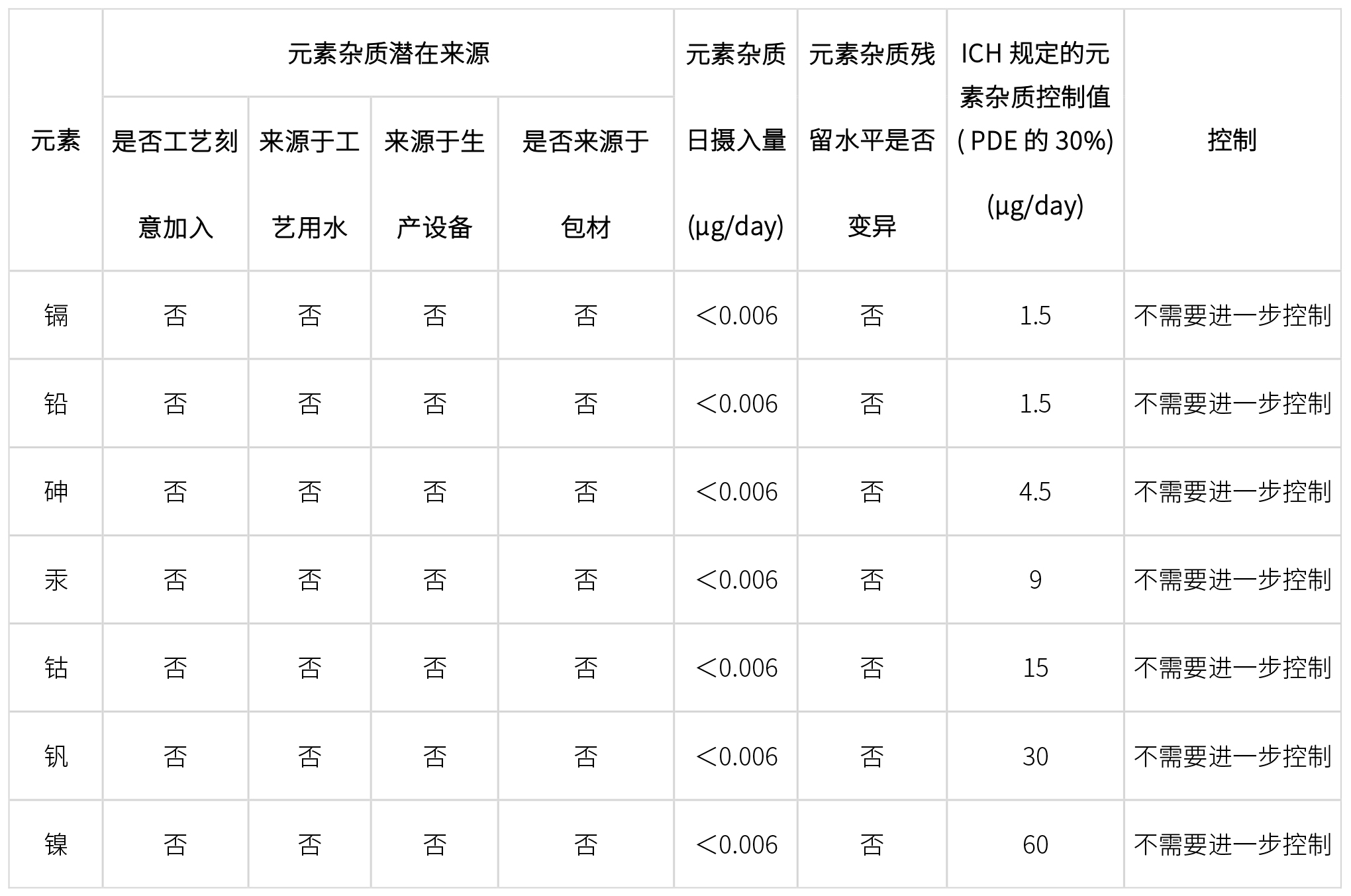
原料药元素杂质控制需要进行风险评估,主要从以下6个方面进行风险评估确定杂质控制策略,如下:
-
起始原料生产工艺引入
-
API生产工艺引入
-
生产设备引入
-
工艺用水引入
-
包材引入
-
元素杂质变异性评价(三批检测数据)
经过上述一系列风险评估和相关检测数据,确定原料药中元素杂质最终控制策略:
➯ API的质量标准
根据中国药典格式,包括性状、鉴别、检查和含量。
-
性状:包括外观,味道,溶解度,熔点,比旋度等
-
鉴别:至少有两个专属性鉴别,包括IR鉴别,UV鉴别,HPLC鉴别,化学鉴别
-
检查:有关物质,异构体,残留溶剂,水分,干燥失重,炽灼残渣,重金属,氯化物,硫酸盐等(除性状,鉴别,含量外其他所有项目)
-
含量:含量通常不小于98.5%(中国药典没有规定上限,默认101.0%)
API质量标准,应参照所有相关药典,检测项目覆盖所有药典,检测限度选取药典最严格的限度制定,如下:
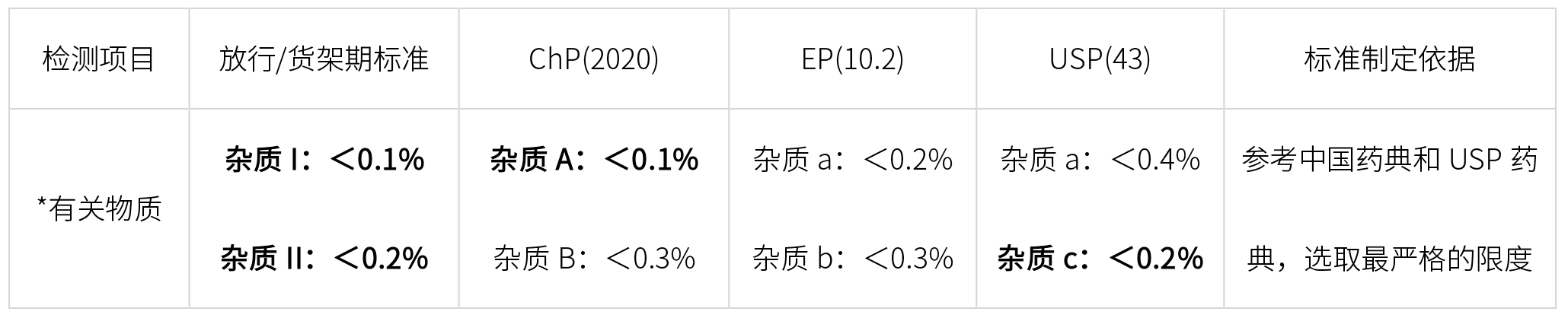
*特定杂质,药典杂质编号可能不一样,进行对比研究时,要一一对应。
标准原料药的质量标准(加粗必有项目)

(*加粗字体,标准中必须包含项目,常规字体根据实际检测结果定是否增订入质量标准)
通过EMA审评报告或IF文件,可以了解包材和稳定性相关信息,主要对光是否敏感,对热是否敏感,是否具有吸湿性等。
稳定性考察内容:影响因素考察(包括包材研究)
-
光,在白炽灯和UV灯照射下,总照度不低于1.2×106Lux·hr,UV不低于200w·hr/m2。放置11天,在0、5、11天取样。若前期调研,对光敏感,需要进行多种放置条件,带包装和裸放,确定对光是否敏感,证明包材合理性
-
热,高于加速温度10°C,放置30天,在0、5、10、30 天取样,裸放
-
湿,在25°C,90%±5%,放置30天,在0、5、10、30天取样,裸放,若前提调研产品具有吸湿性,带包装和裸放,确定是否具有吸湿性,证明包材合理性
➯ 稳定性考察内容-加速和长期稳定性(常温条件下)
批次要求:采用注册批次三批;包装:市售包装一致
加速: 40±2°C,75%±5%;中间:30±2°C,65%±5%
长期:25±2°C,60%±5%
取样频率:加速:0、1、2、3、6;长期:0、3、6、9、12、18、24、36、48
监控项目:加速6个月,长期12、24、36、48个月需要进行全检;
其他项目可根据稳定性变化项目进行考察
显著变化:对于API来说,超出限度,就是显著变化
➯ 稳定性考察内容-加速和长期稳定性(冷冻条件下)
批次要求:采用注册批次三批;包装:市售包装一致
长期:-20±5°C 取样频率:长期:0、3、6、9、12、18、24、36、48
监控项目:长期12、24、36、48个月需要进行全检;其他项目可根据稳定性变化项目进行考察
备注:了解短期偏离储藏条件(运输或搬运过程)对API影响,即采用一批原料药,在略高温度(5±3°C/25±2,60%±5%),放置适当时间(例如10天,20天或1个月)且取样更频繁的测试论证
参考文献
ICH Q7 活性药物成分(API)的GMP指南
ICH Q3A 新原料药中杂质
ICH Q3C 残留溶剂的指导原则
ICH Q3D 元素杂质指导原则
EMA残留溶剂指南
EMA关于沙坦药品中亚硝胺风险评估报告
80号文化学药品新注册分类申报资料要求
-END-
关于我们:
原料药研究事业部现有研发人员近百人,近80%为硕士或博士学历。由具有10年以上国内外项目管理、药品开发、生产转化、注册申报经验人员作为主要研究和管理者,形成具有集产品立项、高端中间体、工艺开发、质量全面研究、产业化转移、中美双报、单制剂原料药评价于一体的原料药全产业链构架。
事业部由6大中心组成,分别是工艺研发中心、产业化中心、质控中心、杂质中心、注册中心和技术服务中心。工艺研发中心主要包含抗生素研发平台、创新工艺研发平台、复杂药物分析研发平台、结晶研发平台和原料药粉体学研究平台。产业化中心主要包含起始原料、API资质控制平台、工艺验证、技术转移风险控制平台。质控中心主要包含方法学建立和验证平台、杂质研究技术包服务平台。杂质中心主要包括杂质制备、MS检测和公共对照品控制等。注册中心注册经验丰富,可以承接中美双报。技术服务中心服务于立项、市场、制剂、采购部等与原料相关的技术支持。
新领先医药原料药事业部可以助力广大业内中间体生产商实现产业升级为原料药供应商、助力制药企业快速确定起始原料供应商、快速完成研发、转移、注册申报,获得生产批件。


转载声明:未经本网或本网权利人授权,不得转载、摘编或利用其他方式使用上述作品。已经本网或本网权利人授权使用作品的,应在授权范围内使用,并注明“来源:新领先医药科技”。

 010-61006450
010-61006450 联系地址:
联系地址: 技术市场部:
技术市场部: 010-61006450
010-61006450
