 Hotline服务热线:010-61006450
Hotline服务热线:010-61006450
 简体中文
简体中文敲重点!QC在药品研发上市过程中的重要作用
按照最新版《药品管理法》(2020版)要求,在药品全生命周期中需执行四个最严,即“最严谨的标准、最严格的监管、最严厉的处罚、最严肃的问责”。为保证药品的安全性和有效性,药品研发作为药品生命周期的源头,需在遵循《药品管理法》的前提下执行好《药品注册管理办法》法规要求。药品研发按创新药、改良型新药、仿制药进行分类,根据不同的注册分类对药品新项目的立项、申报临床、申报生产、批准上市有着不同的资源配置要求。
在“化学药品注射剂仿制药质量和疗效一致性评价技术要求”中提出[1]“注射剂稳定性研究的加速试验、长期试验应在符合 GMP条件下进行”,体现出“完善的质量控制(QC)和质量保证(QA)体系”对药品研发的重要性,现通过以下几个方面对QC(质量控制quality control)在药品研发中的重要性进行阐述与探讨。
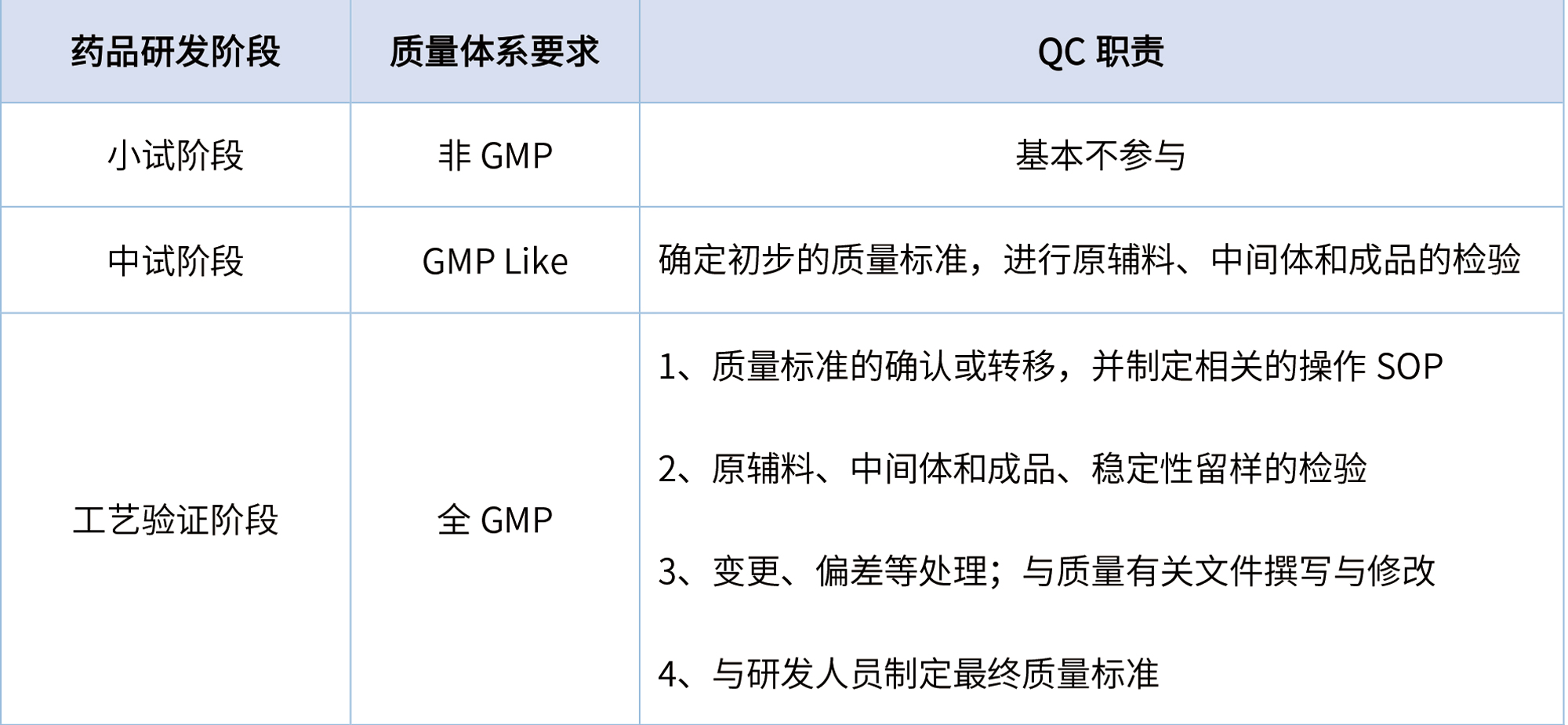
从药品研发开始至批准上市,按GMP体系运行要求,可分为非GMP阶段、GMP Like阶段和全GMP阶段,QC人员需参与的具体工作见表1。
表1 药品研发各阶段QC的职责
QC人员需具备较强的药物分析专业知识、实验操作能力和质量意识,在研发人员的指导下,运用QbD的理念参与到药品研发中,以提高检验检测的效率,并减少因检验检测产生的异常、偏差而导致的生产工艺无法重现、产品质量无法保证的问题。
药品质量标准最终需由符合GMP条件下的检测实验室确认、复核、定稿。现从仪器与量器的确证、方法的确认、人员的管理三个方面阐述检测实验室对质量标准确定所起的作用和需QC人员完成的工作。


-
仪器与量器的确证
GMP条件下检测实验室对仪器设备的采购、安装、验收、运行、维护和记录均有严格要求;《检测和校准实验室能力认可准则》(CNAS-CL01:2018)6.4设备项下要求对仪器设备的“处理、运输、储存、使用、按计划维护、校准和期间核查”应按程序文件执行。
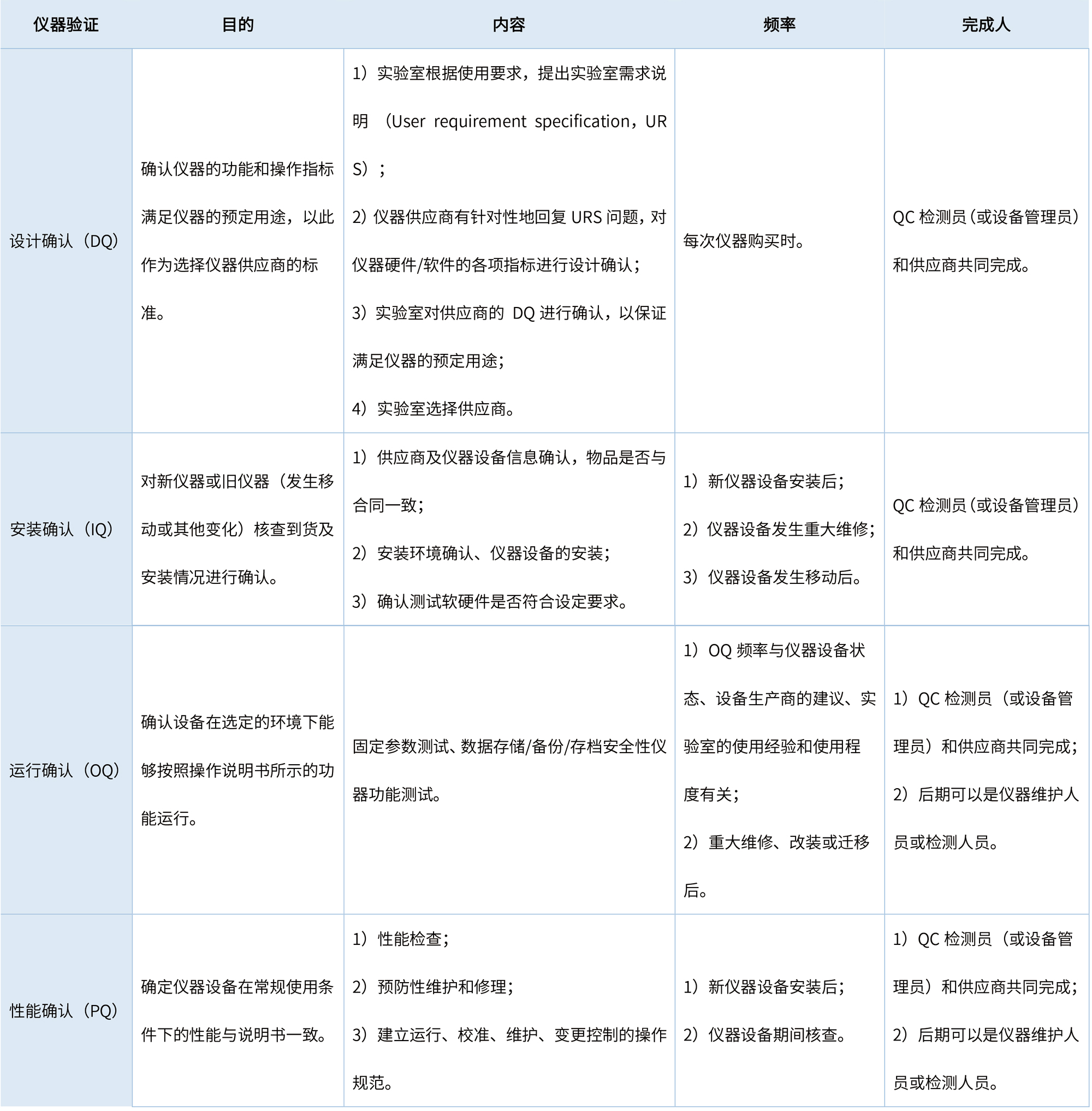
仪器运行之前的一系列工作可称之为“仪确证”[2],由GMP规范和《检测和校准实验室能力认可准则》可知,“仪器确证”工作是检测实验室质量保证的重要途径,需QC相关人员配合完成对应的实验和文件。仪器确证是实验室对仪器进行的生命周期全过程管理,通常用4Q确认的方式来体现,即DQ、IQ、OQ、PQ,具体内容见表2。
表2 仪器4Q确认
在方法学验证、确认、转移过程中所使用的仪器设备、玻璃量器均需通过计量校准或检定,因为所用工具和仪器的精度直接决定检验结果的精度。仪器设备除按周期进行校准外,还需进行期间核查和仪器设备的维护保养,以保证仪器设备的正常运行、检测数据的准确性和可靠性。仪器设备、玻璃量器的内部校准、维护保养均由受培训过的QC相关人员完成。
-
方法的确认
准确、可靠的方法是获得可靠性数据的前提,是保证药品研发过程中产品生产工艺稳定、成品质量的重要质控手段。分析方法的确认[3~4](verification)和转移[5~6](transfer)是检测方法生命周期中的重要环节,两者都是对已验证[2,7](validation)方法是否能持续满足其预期用途的确证过程。
在方法验证阶段,需要对实验人员、仪器和主要试剂等主要变异源进行多因素的全面联合评价,属于多因素的正交设计或嵌套设计。而方法确认或转移,不需要再进行这种全面的多因素设计,仅需实验人员在规定条件下独立重复即可达到确认评估目的[8]。
目前国内有关方法确认/转移的指导原则中,通常是简单比较结果的平均值和偏差(例如中间精密度的计算),以此作为对方法确认/转移成功判定的评价参数[9],这种方式未从整体体现方法的变异,也没有直接回答方法是否满足其预期用途。
《中国药典》2020版四部目前采用对准确度和精密度单独考察的方式,割裂了二者之间的关联,无法准确地对方法的性能是否满足其预期用途进行判断;USP<1210>[10]采用预测区间、容忍区间和方法能力指数(MCI)这些参数作为方法确认/转移的评估标准,可更全面地了解方法的性能。
由上述分析可知,分析方法的验证、确认和转移时,评估标准非常重要;同时,人员操作的熟练程度和规范性、仪器设备的灵敏度、计算化系统等质量体系要素决定了该方法在生产企业QC实验室转移是否成功,是否可以保证后期工艺验证的质量可靠性。
-
人员的管理
检测员是实验室数据质量的重要保障人员,QC实验室对每位入职检测员进行质量体系规范性相关的文件、专业知识、操作技能等多维度的培训,每项操作通过理论考试和现场操作考试后,获得上岗证方可进行实验操作。
GMP条件下检测实验室进行实习检测员监督、在岗人员监控、实验室间比对、内部和外部检测能力监控,以保持检测能力的持续性和可靠性。与其比较,研发岗位对实验员要求做得不如QC人员严格,研发人员缺乏文件意识、质量与风险意识、PDCA理念处理问题的能力及持续改进的能力。
上述3个条件是QC实验室准确复核即将上报“国家食品药品监督管理局”质量标准的前提。
-
QC是新产品申报生产、批生产前一系列活动完成的主体
在申报生产前,产品的中试批及工艺验证,需要QC人员确定检测方法、参与检测,配合完成偏差和异常数据处理,出具中间体检测报告、批全检报告等。工艺验证结束后,需按国家食品药品监督管理局要求,在GMP条件下进行稳定性留样和检测,为申报生产提供数据。
新药或仿制药品上市前,国家食品药品监督管理局确认相关生产和质量控制活动与申报的处方、生产工艺、生产条件、质量标准的一致性,安排对所受理药品注册申请批准上市前的样品批量生产过程,此动态核查的过程需QC部门配合中控检测,为生产出合格产品提供依据。
-
QC是药品上市前后验证工作必不可少的重要参与者和完成者
药品上市前后需进行持续验证,如厂房、工艺、清洁等全方位的验证,以保证新产品在GMP车间的顺利投产。这些必须由QC进行实时跟进并完成相关的检测,并出具报告。
药品上市前后计划性、非计划性验证,需QC人员配合完成相关的检测。计划性的验证包括:总计划执行、产品验证、运输验证、包装验证等;非计划性验证包括:供货及生产等异常验证、不同包装形式的验证、生产过程等待验证,客户反馈的应急性验证、风险和变更识别时的主动验证、偏差投诉发生时的补充性验证。上述验证均需要质量合规意识强、业务能力优秀、实践经验丰富的QC人员用QbD理念制定方案、实时参与检测、识别风险和机遇、判断异常数据和偏差,以得到准确、可靠的检测结果,服务于生产与市场。
-
QC是药品上市后产品质量生命周期的重要践行者
药品上市后工艺验证批次、重要批次的稳定性留样后所有的检测工作需QC人员完成,将数据提供给相关部门进行必要的补充申请,如延长药品的有效期。
药品上市后所有批次的中间体控制和成品检测均由QC完成,QC可根据实际检测情况进行产品质量标准提高。
QC为上市后药品年度报告提供了客观的数据。
-
QC是质量体系维持与持续改进、预防性验证的主要执行者
QC在完成大批量的原辅料检测、药品批检验的过程中,积累了大量经验,使其具有扎实的药品检验理论知识和较强的实践操作能力,有足够能力识别检测中存在问题的关键点和潜在的风险,有能力进行预防性管理和事前的警示。通过差异性识别和分析,结合生产过程的实时重现和验证,可以避免系统问题和严重问题的发生。
由以上可知,QC是药品成功上市的重要桥梁,检测实验室在遵守《药品生产管理规范(2010版)》基础上[11],通过CNAS-CL01:2018、PQ等各项认可准则,更好地服务于药品的研发和生产。
在2015~2019年,FDA 对我国药企发出77封警告信,约占其全部警告信数量的20% ,共263条违背 cGMP 的缺陷项,其中质量控制与质量保证问题72条占27.4%,仅次于“物料与产品”[12],主要问题包括:
-
未制定药品稳定性试验管理相关文件,药品有效期或复验期缺少适当的稳定性试验支持;
-
未能对计算机系统实施充分的控制,以阻止未经授权的访问、修改及删除数据,不能保证相关数据的可靠性,无法确保药品符合既定的质量标准。
这两个问题的存在,说明有些企业在QC实验室的合规性建设方面仍有缺陷。QC实验室在遵守ISO/IEC 17025准则的基础上,应积极借鉴制药企业GMP管理理念和要求,完善QC实验室在记录与数据可靠性、变更控制和偏差调查等方面的体系建设,主动提升质量管理水平,提高数据可信度,保证更多产品在FDA和欧盟上市,提高国际市场竞争。
参考文献:
1、国家药品监督管理局药品审评中心,化学药品注射剂仿制药质量和疗效一致性评价技术要求(2020年第2号文),https://www.cde.org.cn/main/news/viewInfoCommon/d9c6f118b773f54e8feba3519bf78a11.
2、CNAS-GL040:2019《仪器验证实施指南》[EB/OL].https://www.cnas.org.cn/rkgf/sysrk/rkzn/2019/09/899950.shtml.
3、USP 41-NF 36[S].2018:7671<1226>
4、中华人民共和国药典2020 年版.四部[S].2020:478<9099>
5、USP 41-NF 36[S].2018:7663<1224>
6、中华人民共和国药典 2020 年版.四部 [S].2020:479<9100>
7、中华人民共和国药典 2020 年版.四部 [S].2020:480<9101>
8、李娜,耿颖,谭德讲,等. 分析方法确认和转移的评估标准探讨[J].药物分析杂志,2021,41(4):675.
9、许明哲,黄宝斌,杨青云,等.分析方法转移内容介绍[J].药物分析杂志,2015,35(1):176.
10、USP 41-NF 36[S].2018:7622<1210>
11、国家食品药品监督管理局. 药品生产质量管理规范 [S]. 2011.
12、颜若曦,曹轶,董江萍.FDA 对我国药品生产企业检查分析[J].中国新药杂志,2020,29(15):1697.
-END-
关于我们:
北京新领先(股票代码:600222)成立于2005年,是一家面向全球提供药学临床前研究、临床CRO和CDMO服务的高新技术企业,连续多年被评为“中国医药研发公司10强(2019年位列第一)”。公司总部位于北京中关村高新技术园区,同时在郑州临空生物园区建立了新药筛选及检测平台、药物评价平台(动物房,GLP、AAALAC、CNAS认证)、大分子中试及大规模生产服务平台、小分子CMC制剂研究生产平台、细胞技术服务平台和临床CRO平台等六大符合国际标准(FDA、EMA和NMPA GMP标准)的研发平台,形成“新领先CXO”全产业链服务体系。仿创结合,双引擎驱动,能够为客户提供药学研发全生命周期的多元化服务。


转载声明:未经本网或本网权利人授权,不得转载、摘编或利用其他方式使用上述作品。已经本网或本网权利人授权使用作品的,应在授权范围内使用,并注明“来源:新领先医药科技”。

 010-61006450
010-61006450 联系地址:
联系地址: 技术市场部:
技术市场部: 010-61006450
010-61006450
