 Hotline服务热线:010-61006450
Hotline服务热线:010-61006450
 简体中文
简体中文专题分享 | 药品生产设备清洁残留限度确认及分析方法验证
药品生产后必须对其生产设备采取一定的程序进行清洗,防止药物成分残留到下一批产品中,形成交叉污染,从而影响下一批产品的质量及安全性。清洁验证是证实清洗程序合理性的必要技术手段。
WHO、EU、ICH、FDA及我国药品GMP均对药品生产清洁验证提出了相关要求:
1)使用同一生产设备生产不同的产品,必须进行清洁验证,确定可接受药物残留水平。
2)当同一设备仅生产一种产品时,不涉及交叉污染,无需开展系统的残留物限度清洁验证,通常通过目视法检查设备表面无可见残留物并保障微生物水平即可。
清洁验证的取样方法主要有擦拭取样法(直接取样)和淋洗取样法(间接取样),为达到清洁目的,两种方法可结合使用。通常根据待清洁品种各组成成分的清洁难易程度、溶解度和允许最大残留量(MACO值)来确定需要清洁的组分。
本文参照欧洲原料药委员会(APIC)在原料药工厂清洁验证指南(2016.09)中给出的药物活性成分残留限度的确定方法,结合国内清洁验证限度确定相关文献,对药品生产设备清洁中活性成分残留限度确认方法进行简要归纳,并初步拟定待清洁活性成分的分析检测方法验证方案。
(清洁验证的接受标准)
在可以获得可接受日暴露水平(ADE) 或允许日暴露量(PDE) 值时 ,可采用ADE或PDE值计算最大允许残留量(MACO)。
首先,计算ADE值或PDE值,计算公式如下:
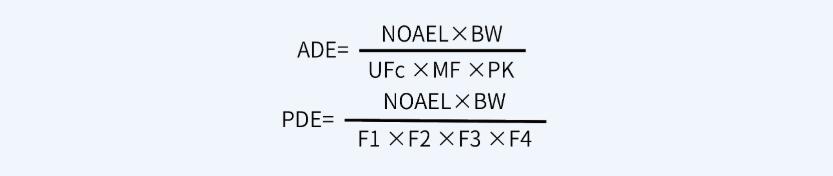
其次,根据ADE值或PDE值计算MACO值,计算公式如下:

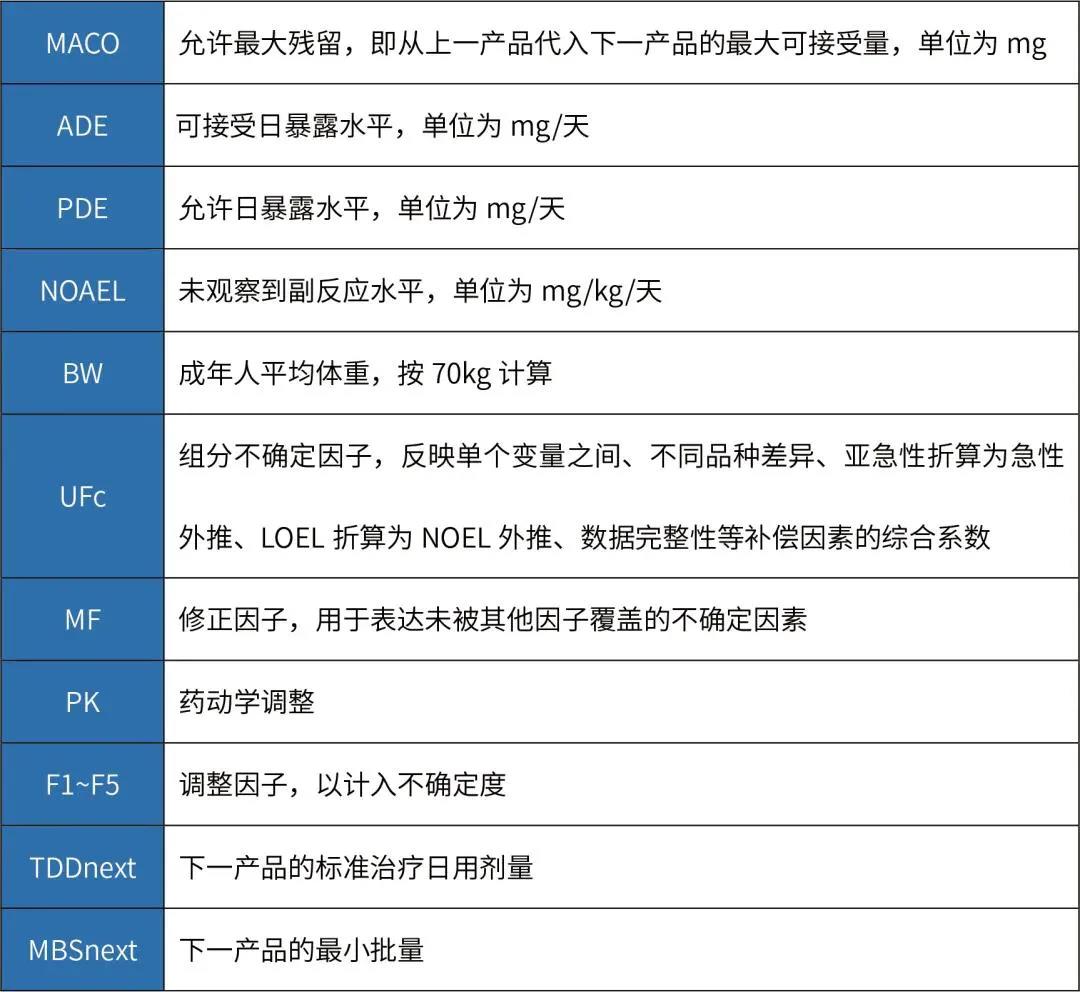
当多品种共线生产时,可以选择ADE或PDE值较小的品种作为上一产品,选择MBS/TDD比值最小的品种作为生产的下一产品。
如果可以获得有限毒性数据和日治疗剂量(TDD)值,可以采用本计算方式,就原料药而言,该公式适用于原料药生产工艺A更换到原料药生产工艺B的情况。计算公式如下:
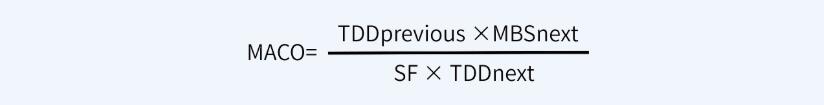

如果无法获得其它数据(例如, ADE、 OEL、 TDD等值),仅能获得半数致死量数据(例如化学物质、中间体、清洁剂等), MACO可以基于半数致死量数据来计算 。
首先,计算NOEL值(无可见影响水平):
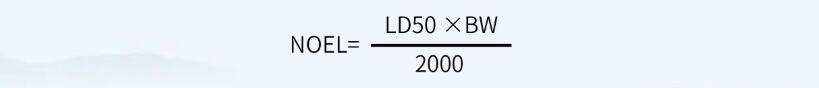
其次,根据NOEL值计算MACO值:
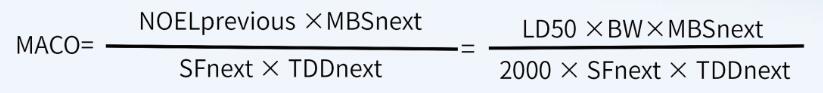
可利用以下公式,基于一个通用限度确定MACO限度,单位为ppm。
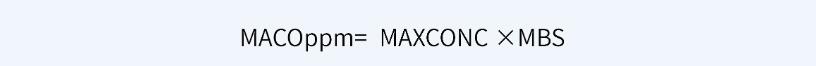
例如,就原料药而言,通用限度为100ppm时,MACO为最小批量(MBS)的0.01%;通用限度为10ppm时,MACO为最小批量(MBS)的0.001%。
通过上述公式,确定MACO值后,即可通过如下公式,计算擦拭法及淋洗法的残留可接受标准。
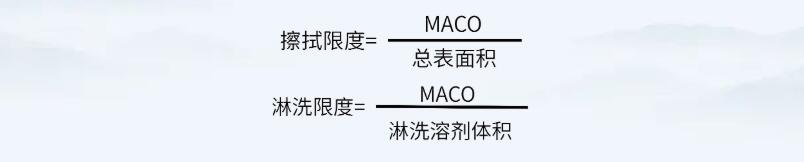
注意,上述公式中擦拭限度计算的“总表面积”可为共线产品共用设备的累积面积。
另外,关于一般限度10ppm,即10μg/ml或10mg/kg,对于液体制剂设备的清洁验证标准确定,淋洗法限度即为10μg/ml。设下一批产品的生产批量为B(ml或kg),上批产品与下批产品的设备共用累积接触面积为D(cm2),为确保安全,应除以安全因子F(可依据药物生理活性水平确定,通常分为1000、100、10三个级别),可按照如下过程计算擦拭法残留限度Ld。
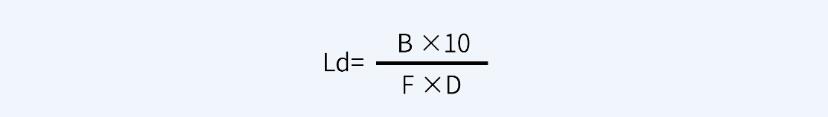
根据各公司所生产产品的属性不同(例如,毒性、药物活性等),从上一产品带入下一产品中的污染物质最大浓度通用上限通常设定为5~500ppm(原料药设定为100ppm较为常见,注射剂可设定为10ppm)。
以上主要给出了4种药物残留限度的计算标准,其中第一种(基于健康数据)及第三种(基于半数致死量)方法更为科学,但因ADE、PDE值或LD50值难以明确,故用之较少,而第二种(基于日治疗剂量)及第四种(基于一般限度)方法更为常用。因此,在确定残留限度时,应综合评比上述几种方法,基于最大程度上降低交叉污染的原则,选择最为合适的结果作为清洁验证的最终可接受标准。
在正式开展清洁验证之前,需进行针对性分析方法的开发与验证。通常选择灵敏度较高的方法,如高效液相色谱法、气相色谱法、紫外分光光度法等。首选可定量的检测方法,也可选择限度检测方法。一般情况下,可以直接选择待清洁品种质量标准中规定的含量测定方法。所选用的分析方法均需按照《中国药典》(2020年版)四部通则9101 分析方法验证指导原则进行验证。
制剂清洁验证分析方法主要验证项目及可接受标准如下:
擦拭法清洁验证分析方法验证
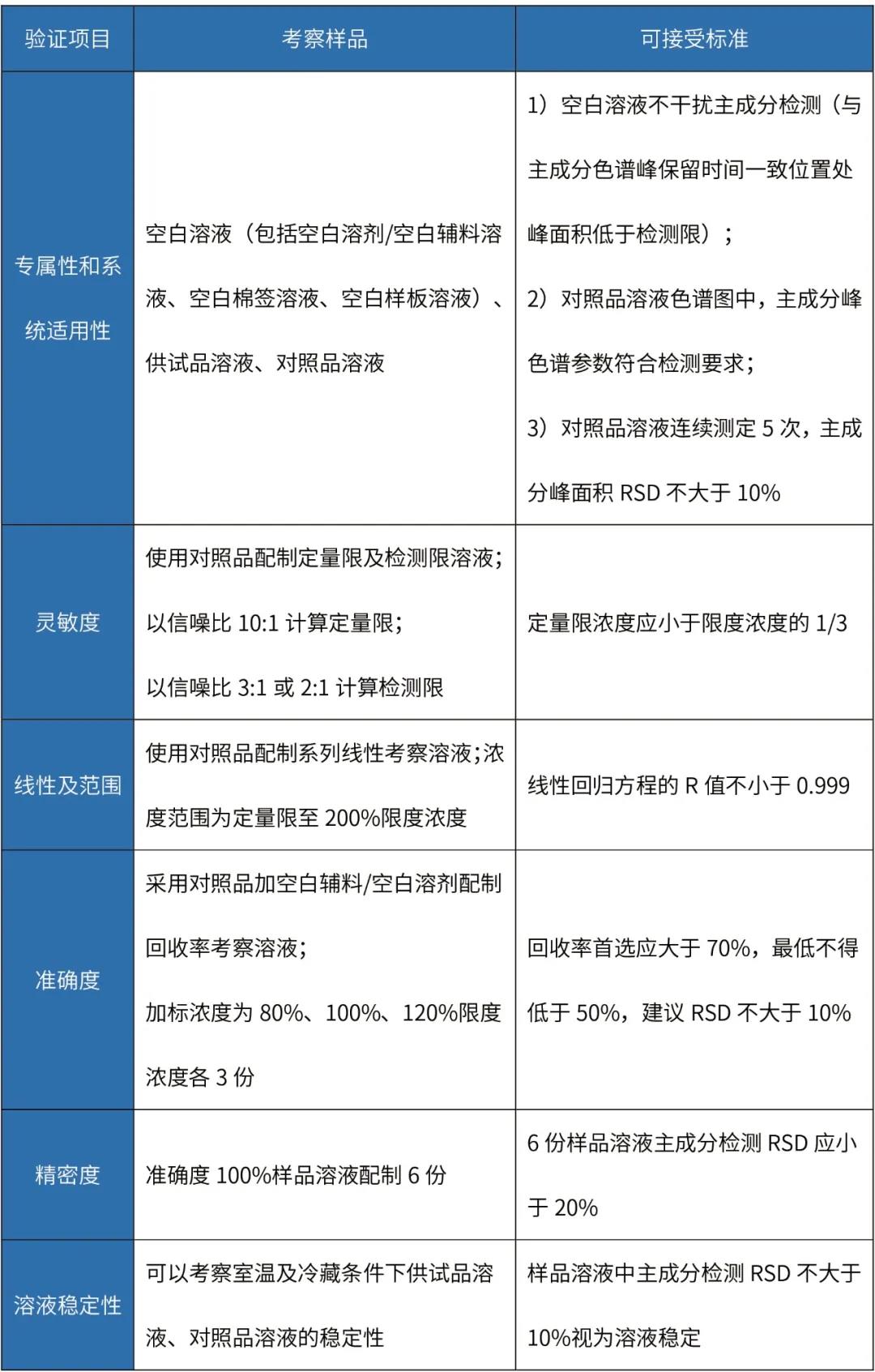
备注:
-
擦拭法准确度试验样品回收过程:将配制好的回收率考察溶液,定量添加至取样板表面(根据设备情况确定取样板材质,规格通常为5cm×5cm),使取样板风干,按照拟定方法用棉签擦拭取样板表面,照供试品溶液制备方法制备样品溶液。
-
擦拭取样操作方法:用溶剂润湿药签,并将其靠在溶剂瓶边挤压,以去除多余的溶剂。将药签头按在取样表面上,用力使其稍弯曲,平稳而缓慢地擦拭取样表面,在向前移动的同时,将其从一边移动到另一边。擦拭的过程应覆盖整个取样表面,然后翻转药签,让其另一侧也进行擦拭,但其方向应与前次垂直。
擦拭法取样药签擦拭方法示例如下图所示:
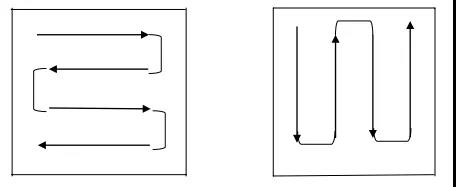
药签擦拭取样示意图
表 淋洗法清洁验证分析方法验证
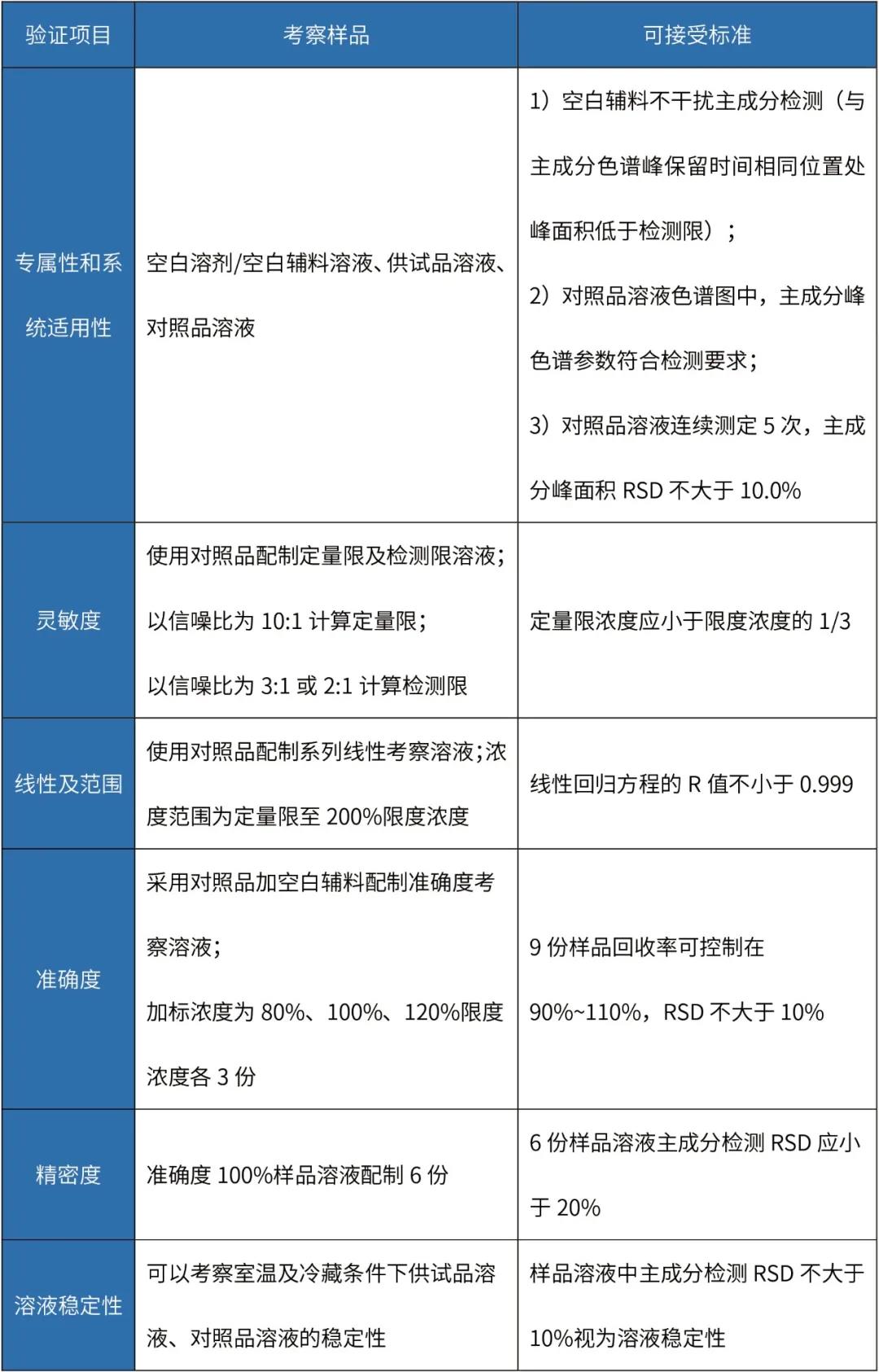
注:该表中淋洗法准确度试验考察的是检测方法的准确度,并非淋洗回收率,接受标准可依据限度酌情调整。
A注射液清洁验证残留限度确认过程如下:
无法获得该注射液有效成分的ADE值或PDE值。
全部共线产品中,B注射液的日用剂量最大,折算成体积为24ml,其最小生产批量为200L;共用设备累积接触面积为367380cm²。
✎ 计算方法一:基于日治疗剂量的可接受标准(先生产A产品,再生产B产品)

其中:
-
MACO代表产品A能进入到产品B中的最大可接受量(mg);
-
MBS代表B产品的最小批量(ml或g);
-
TDDprevious代表A产品的日最小治疗剂量(mg);
-
TDDnext代表在设备中生产B产品的最大日剂量(ml或g);
-
SF代表安全系数,可取1000。
注:MBS与TDDnext单位应统一。
对于本产品而言,产品A最小治疗剂量按50mg计,产品B最小批量200000ml,最大日剂量为24ml。因此A注射液的有效成分能进入到B注射液中而不致引起风险的最大量为:
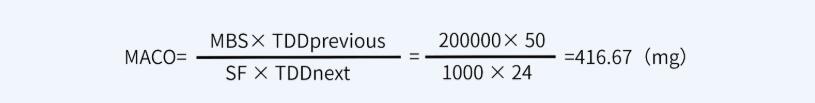
淋洗水按200L计,则淋洗水残留限度416.67mg÷200L=2.083μg/ml
共用设备累积接触面积为367380cm²,
则擦拭残留限度为416.67mg÷367380cm²=1.134μg/ cm²
✎ 计算方法二:基于半数致死量的可接受标准(先生产A产品,再生产B产品)
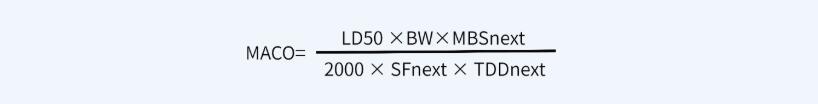
其中:
-
MACO代表产品A能进入到产品B中的最大可接受量(mg);
-
MBS表产品B的最小批量(ml或g);
-
TDDnext代表产品B的最大日剂量(ml或g);
-
SF代表安全系数,一般取1000;
-
LD50代表动物的半数致死量,证明动物(如小鼠)和给药途径(如静注\口服)很重要(mg/kg);
-
BW代表一个成年人的平均体重,按70kg计算;2000代表一个经验常数。
注:MBSnext与TDDnext单位应统一。
对于本产品而言,产品A的小鼠半数致死量按照156mg/kg(静脉注射)计,产品B的最小批量为200000ml,最大日剂量为24ml。因此A注射液的有效成分能进入到B注射液中而不致引起风险的最大量为:
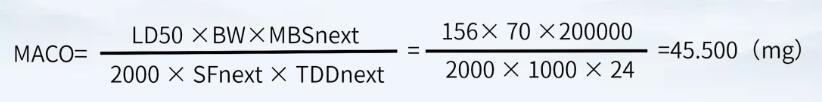
淋洗水按200L计,则淋洗水残留限度
45.500mg÷200L =0.228μg/ml
共用设备累积接触面积为367380cm²,
则擦拭残留限度为45.500mg÷367380cm²=0.124μg/ cm²
✎ 计算方法三:一般限度,采用上批产品对下批产品的污染不得超过10ppm(10mg/kg)作为限定指标
最小批量200L,共用设备累积面积为367380cm²
淋洗法限度:10ppm=0.001%=0.001g/100ml=10μg/ ml
擦拭法限度:

其中:
-
B代表B产品的最小批量(ml);
-
D代表共用设备累积面积(cm2);
-
F代表安全因子,一般取10。
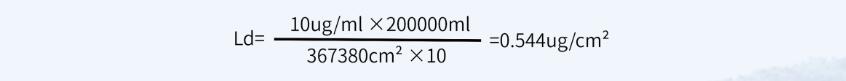
采用上述3种计算方法中结果最小的限度值,即擦拭法残留限度不得过0.124μg/ cm²,淋洗法残留限度不得过0.228μg/ml。
清洁验证是防止药品生产过程中发生污染与交叉污染的重要措施之一,企业应针对药品特性、生产设备特点等因素建立明确可控的清洗程序,并依据科学分析制定合理的残留限度标准,建立并验证科学的取样及分析方法,以确保验证结果准确,药品生产稳定可控,药品质量安全可靠。
-END-
关于我们:
北京新领先(股票代码:600222)成立于2005年,是一家面向全球提供药学临床前研究、临床CRO和CDMO服务的高新技术企业,连续多年蝉联“中国医药研发公司榜首”。公司总部位于北京中关村高新技术园区,同时在郑州临空生物园区建立了新药筛选及检测平台、药物评价平台(动物房,GLP、AAALAC、CNAS认证)、大分子中试及大规模生产服务平台、小分子CMC制剂研究生产平台、细胞技术服务平台和临床CRO平台等六大符合国际标准(FDA、EMA和NMPA GMP标准)的研发平台,形成“新领先CXO”全产业链服务体系。仿创结合,双引擎驱动,能够为客户提供药学研发全生命周期的多元化服务。


转载声明:未经本网或本网权利人授权,不得转载、摘编或利用其他方式使用上述作品。已经本网或本网权利人授权使用作品的,应在授权范围内使用,并注明“来源:新领先医药科技”。

 010-61006450
010-61006450 联系地址:
联系地址: 技术市场部:
技术市场部: 010-61006450
010-61006450
